
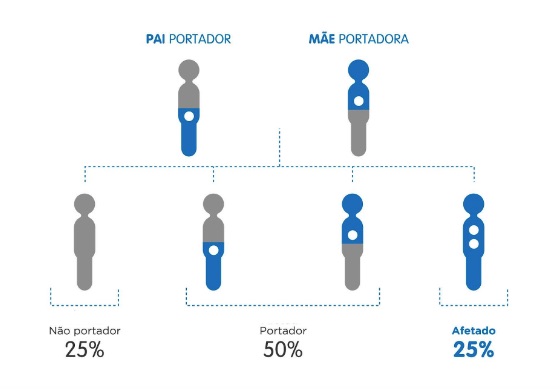
A fibrose cística é uma doença genética causada por alterações no gene chamado CFTR (Cystic Fibrosis Transmembrane Regulator), que gera um desequilíbrio na concentração de cloro e sódio nas secreções do corpo, como muco e suor (glândulas exócrinas). A doença não pode ser adquirida ao longo da vida, ou seja, para que alguém tenha fibrose cística é necessário que nasça com ela. Uma criança não “pega” e nem “transmite” a FC. Tanto a mãe como o pai geralmente carregam o gene afetado pela doença, ainda que ambos não apresentem os sintomas da fibrose cística. Apenas quando a criança recebe as cópias dos genes com mutação dos seus pais ela vai manifestar a doença, pois terá duas cópias com mutações no gene CFTR (figura 1). Assim é importante pontuar que se um casal gerou um filho com FC, existe uma grande possibilidade (25%) de um novo filho nascer com a doença.
Como é feito o diagnóstico da Fibrose Cística?
O diagnóstico da fibrose cística é estabelecido quando um pessoa com sintomas sugestivos ou um teste de triagem neonatal alterado tem duas dosagens de cloro no suor alteradas (>60mEq/L) ou duas mutações causadoras de fibrose cística identificadas no teste genético.
- Triagem neonatal (teste do pezinho): a triagem neonatal consiste na retirada de algumas gotas de sangue do calcanhar do bebê, na primeira semana de vida. O teste de triagem neonatal não é um exame definitivo, ou seja, ele não determina se o bebê tem ou não fibrose cística, mas é capaz de indicar a necessidade de exames complementares para definir se o indivíduo tem de fato a doença. A realização do teste do pezinho é fundamental, pois permite realizar o diagnóstico antes do aparecimento dos primeiros sintomas da fibrose cística, melhorando muito o prognóstico. Além disso, o diagnóstico precoce possibilita o aconselhamento genético aos pais e familiares em relação a futuras gestações. O teste do pezinho também investiga uma série de outras doenças potencialmente graves.
- Teste do suor: é realizado apenas quando há suspeita da fibrose cística, através da indução e coleta do suor. O teste avalia a concentração de cloro (ou cloreto) no suor, que acima de um determinado nível indica a existência da doença (60 mEq/l ou mais, dependendo da técnica utilizada).
- Teste genético: é uma opção para o diagnóstico de pessoas com suspeita de fibrose cística que apresentam níveis intermediários de cloreto no suor (entre 30 e 60 mEq/l). Além disso, é indicado para determinar o genótipo das pessoas com fibrose cística, podendo auxiliar no aconselhamento familiar e determinar possibilidades de tratamento com novos medicamentos que “corrigem” a função da proteína CFTR.
Quais são as alterações que um bebê ou uma criança com FC podem apresentar?
Entre as consequências da fibrose cística está a produção de muco exageradamente espesso, que não é devidamente eliminado pelo organismo. O acúmulo do muco em órgãos como pulmões, pâncreas, fígado e intestino – provoca alterações no funcionamento dos mesmos.
Nos pulmões, este acúmulo de secreção leva ao aparecimento de tosse com catarro e favorece o aparecimento de bactérias, que causam infecções graves e constantes, causando inflamação pulmonar e colocando a saúde das pessoas em risco.
O pâncreas é responsável pela produção das enzimas digestivas, que são substâncias que quebram os alimentos para que eles possam ser absorvidos pelo corpo. Na fibrose cística, o entupimento dos canais do pâncreas impede que as enzimas cheguem ao intestino e a maior parte dos nutrientes acaba se perdendo nas fezes. Esta dificuldade de absorver os alimentos pode atrapalhar o ganho de peso e crescimento, especialmente nos bebês. A nutrição é um ponto muito importante para a saúde das pessoas com fibrose cística.
Outra característica importante da pessoa com fibrose cística é o suor mais salgado, podendo levar ao aparecimento de cristais de sal na testa das crianças menores. Nos dias de calor, esta perda de sal no suor pode levar até a uma desidratação grave, mesmo sem diarréia ou vômitos.
É importante ressaltar que as pessoas com fibrose cística têm um excelente potencial para ter uma vida plena, com desenvolvimento totalmente normal e podendo frequentar a escola, fazer esportes e ter uma vida familiar e social ativa.
Apesar de ainda não existir uma cura definitiva para a fibrose cística, existem vários recursos de tratamento que podem proporcionar uma vida saudável e produtiva. Diversas pesquisas estão em andamento para novos medicamentos que podem melhorar ainda mais a qualidade de vida das pessoas com fibrose cística. Para isso, o diagnóstico precoce e a adesão ao tratamento são fundamentais, possibilitando uma vida saudável e produtiva.



0 comentários